
Creutzfeldt-Jakob Disease (CJD)
Prion diseases have long incubation periods and progress inexorably once clinical symptoms appear. Human prion diseases comprise: Creutzfeldt-Jakob disease (CJD or classic CJD), variant Creutzfeldt-Jakob disease (vCJD), fatal familial insomnia (FFI), Gerstmann-Sträussler-Scheinker syndrome (GSS), and Kuru. CJD accounts for more than 90 percent of sporadic prion disease, although it is still rare. CJD occurring in 3 different forms: sporadic CJD (sCJD), familial CJD (fCJD), and iatrogenic CJD (iCJD). The vast majority of CJD cases are sporadic (85 percent), while 10 to 15 percent are due to fCJD; iCJD generally accounts for less than 1 percent.
Sporadic CJD is not thought to be exogenously acquired. Familial CJD, GSS, and FFI are associated with mutations of the prion protein gene and inherited with family. Other prion diseases (iCJD, vCJD, and Kuru) are thought to be acquired exogenously. Humans are the only species affected by the CJD prions in nature, with the exception of vCJD, which represents zoonotic spread of BSE from its reservoir, thought to be in cattle. Subclinical infection with the BSE prion appears to be present in human populations. For acquired forms of CJD (iCJD and vCJD), incubation periods differ from the route of exposure, could range from 15months to more than 30 years. The incubation period for vCJD from consuming BSE-contaminated cattle products is between 10 and 20 years.
Signs and symptoms of CJD are limited to central nervous system dysfunction. Rapidly progressive mental deterioration (often with behavioral abnormalities) and myoclonus are the two cardinal clinical manifestations of sCJD. It typically presents as a rapidly progressive encephalopathy with confusion, dementia, and other neurological signs such as cerebellar dysfunction. Extrapyramidal signs such as hypokinesia and cerebellar manifestations are also common. Clinical phenotypes of sporadic CJD have been associated with molecular subtypes determined by the PRNP gene codon 129 genotype and the pathologic prion protein (PrPSc) type. However, atypical presentations have been reported. Treatment of prion diseases remains supportive; no specific therapy has been shown to stop the progression of these diseases. Death usually occurs within one year of symptom onset.
Epidemiology
Approximately 1 to 1.5 case of sporadic CJD occurs per 1,000,000 population per year with a worldwide distribution, although rates of up to 2 cases per million are not unusual. The risk of CJD increases with age, and in persons aged over 50 years of age, the annual rate is approximately 3.4 cases per million. Sporadic CJD has been reported worldwide with annual mortality rate of 1-2 per million population. The highest age age-specific average mortality rate (>5 cases/ million) occurs in the 65-79 year age group.
Variant CJD typically affects a younger age group than sporadic CJD (median age at death: 28 vs. 68years). The incidence of vCJD cases is falling in recent years.
In 1996, the Taiwan government launched a surveillance program in all the hospitals and sought cases retrospectively back to January 1, 1975. Thereafter, neurologists have been requested to report suspected cases to the government since 1997. If physicians found cases meet the reporting criteria, they should refer patients to the neurological centers for further evaluation. In October 15, 2007, CJD and its variance were announced to be a notifiable disease, category 4 in Taiwan. Over the years, there were 4 fCJD confirmed cases, 1 imported vCJD probable case, 2 biopsy- proven sCJD confirmed cases, others were possible or probable cases of sCJD.
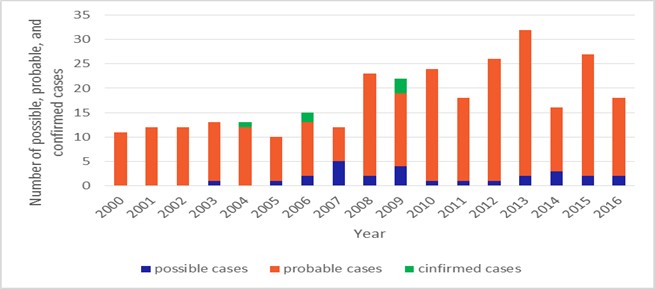
Figure. Number of CJD possible, probable and confirmed cases by Year-Taiwan, 2000-2016.
Creutzfeldt-Jakob Disease(CJD) Surveillance in Taiwan
- Taiwan National Infectious Disease Statistics System–Creutzfeldt-Jakob Disease (CJD)
- Self–reporting through the toll–free 1922 hotline or the local health bureau.
Prevention and Control
- People should never donate blood, tissues, or organs if they have suspected or confirmed CJD, or if they are at increased risk because of a family history of the disease, a dura mater graft, or other factor.
- Avoid reuse of potentially contaminated instruments.
- Avoid organ or tissue transplant from infected patients.
- When management persons with confirmed or suspected prion diseases, specific precautions should be taken. WHO infection control guidelines to minimize the risk of transmission of CJD and sterilization protocols for CJD-contaminated surgical instruments are available.
- Avoid exposures to BSE-causing agent in food bovine origin.
FAQs
- What are the symptoms of CJD?
- CJD is characterized by rapidly progressive dementia. Initially, individuals experience problems with muscular coordination; personality changes, including impaired memory, judgment, and thinking; and impaired vision. People with the disease also may experience insomnia, depression, or unusual sensations.
- CJD does not cause a fever or other flu-like symptoms. As the illness progresses, mental impairment becomes severe. Individuals often develop involuntary muscle jerks called myoclonus, and they may go blind. They eventually lose the ability to move and speak and enter a coma. Pneumonia and other infections often occur in these individuals and can lead to death. These variants differ somewhat in the symptoms and course of the disease. For example, a variant form of the disease called variant CJD (v-CJD), as described in the UK and France, begins primarily with psychiatric symptoms, affects younger individuals than other types of CJD, and has a longer than usual duration from onset of symptoms to death.
- What's the difference between classic and variant CJD?
- The variant form of CJD should not be confused with the classic form of CJD that is endemic throughout the world. There are several important differences between these two forms of the disease. The median age at death of patients with classic CJD in the United States, for example, is 68 years, and very few cases occur in persons under 30 years of age. In contrast, the median age at death of patients with vCJD in the United Kingdom is 28 years. Variant CJD can be confirmed only through examination of brain tissue obtained by biopsy or at autopsy, but a "probable case" of vCJD can be diagnosed on the basis of clinical criteria developed in the United Kingdom. The incubation period for vCJD is unknown because it is a new disease. However, it is likely that ultimately this incubation period will be measured in terms of many years or decades. In other words, whenever a person develops vCJD from consuming a BSE-contaminated product, he or she likely would have consumed that product many years or a decade or more earlier.
- In contrast to classic CJD, vCJD in the United Kingdom predominantly affects younger people, has atypical clinical features, with prominent psychiatric or sensory symptoms at the time of clinical presentation and delayed onset of neurologic abnormalities, including ataxia within weeks or months, dementia and myoclonus late in the illness, a duration of illness of at least 6 months, and a diffusely abnormal non-diagnostic electroencephalogram. The characteristic neuropathologic profile of variant CJD includes, in both the cerebellum and cerebrum, numerous kuru-type amyloid plaques surrounded by vacuoles and prion protein (PrP) accumulation at high concentration indicated by immunohistochemical analysis.
- Is there any treatment or cure for CJD?
- There is no confirmed effective treatment to arrest or cure CJD at the present time. The only treatments available for CJD patients focus on easing their symptoms and discomfort.
- What is being done to protect us from CJD?
- At present there is no specific way of protecting people from developing sporadic or familial CJD. Destroying surgical instruments that have been used on people with CJD and by not using their organs or tissues for transplant guard against iatrogenic CJD. In relation to dietary-related variant CJD, BSE in cattle is being monitored and controlled and there are various measures to protect the human food chain from BSE contamination. There is however remaining concern about secondary transmission of vCJD from individuals who might have subclinical (asymptomatic) BSE/vCJD infection-especially via blood transfusion. However, there are measures in place to reduce any risk and the risk appears to be very low.
- Can you catch CJD from someone?
- Prion diseases are not infectious in the usual way. There is no increased risk of developing CJD from contact with a CJD patient. However, transmission can occur during invasive medical interventions. It is sensible for anyone who might be exposed to the blood of a CJD patient to wear gloves and take standard precautions.
- Is an autopsy necessary in CJD?
- Post-mortem examination is not mandatory in Taiwan.
More
Images

Attached Files
 Government Entry Point」(open in new window)")